

Understanding Nrf2 and its Impact on Neurodegenerative Diseases
Neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, affect millions of individuals worldwide. A variety of treatment options are available to treat the symptoms of several neurodegenerative diseases although the results are often limited. Research studies have found that oxidative stress caused by both internal and external factors can be a cause for the development of neurodegenerative diseases. The transcription factor, Nrf2, has been determined to function as a major defense mechanism against oxidative stress. The purpose of the article below is to show the effects of Nrf2 on neurodegenerative diseases.
Modulation of Proteostasis by Transcription Factor NRF2
Neurodegenerative diseases are linked to the accumulation of specific protein aggregates, suggesting an intimate connection between injured brain and loss of proteostasis. Proteostasis refers to all the processes by which cells control the abundance and folding of the proteome thanks to a wide network that integrates the regulation of signaling pathways, gene expression
Keywords: Neurodegenerative diseases, Unfolded protein response, Proteasome, Ubiquitin, Autophagy, Oxidative stress
Abbreviations
Sciencedirect.com/science/article/pii/S2213231716304050
Introduction
Nuclear Factor (erythroid-derived 2)-like 2 (NRF2) is a basic-leucine-zipper protein considered nowadays as a master regulator of cellular homeostasis. It controls the basal and stress-inducible expression of over 250 genes that share in common a cis-acting enhancer termed the antioxidant response element (ARE) [1], [2], [3], [4], [5]. These genes participate in phase I, II and III detoxification reactions, glutathione and peroxiredoxin/thioredoxin metabolism, NADPH production through the pentose phosphate pathway and malic enzyme, fatty acid oxidation, iron metabolism, and proteostasis [6]. Given these wide cytoprotective functions, it is possible that a single pharmacological hit in NRF2 might mitigate the effect of the main culprits of chronic diseases, including oxidative, inflammatory and proteotoxic stress. The role of NRF2 in the modulation of the antioxidant defense and resolution of inflammation have been addressed in numerous studies (reviewed in [7]). Here, we will focus on its role in proteostasis, i.e., the homeostatic control of protein synthesis, folding, trafficking and degradation. Examples will be provided in the context of neurodegenerative diseases.
Loss of Proteostasis Influences NRF2 Activity in Neurodegenerative Diseases
A general hallmark of neurodegenerative diseases is the occurrence of aberrant aggregation of some proteins. Thus, misfolded protein aggregates of α-synuclein (α-SYN) are found in Parkinson’s disease (PD), β-amyloid (Aβ) plaques and hyper-phosphorylated TAU neurofibrillary tangles in Alzheimer’s disease (AD), huntingtin (Htt) in Huntington’s disease (HD), superoxide dismutase 1 (SOD1) and TAR DNA binding protein 43 (TDP-43) in amyotrophic lateral sclerosis (ALS), prion protein (PrP) in spongiform encephalopathies, etc. Protein aggregates can have an impact on several cellular pathways, which in turn may affect NRF2 levels and activity.
Different Layers of Regulation Tightly Control NRF2 Activity
Under physiological conditions, cells exhibit low NRF2 protein levels because of its rapid turnover. In response to different stimuli, NRF2 protein is accumulated, enters the nucleus and increases the transcription of ARE-containing genes. Therefore, management of NRF2 protein levels is a key point that should integrate positive and negative input signals. As we will discuss further, NRF2 is activated by diverse overlapping mechanisms to orchestrate a rapid and efficient response but on the other hand NRF2 could be inhibited, probably in a second phase, in order to switch off its response.
From the classic point of view, activation of NRF2 has been considered as a consequence of the cellular response to oxidant or electrophilic compounds. In this regard, the ubiquitin E3 ligase adaptor Kelch-like ECH-associated protein 1 (KEAP1) plays a crucial role. Molecular details will be further addressed in Section 4.1. In brief, KEAP1 acts as a redox sensor due to critical cysteine residues leading to NRF2 ubiquitination and proteasomal degradation. In addition to this classic modulation, NRF2 is profoundly regulated by signaling events. Indeed, different kinases have been shown to phosphorylate and regulate NRF2. For instance, NRF2 can be phosphorylated by mitogen activated protein kinases (MAPKs), although its contribution to NRF2 activity remains unclear [8], [9], [10], [11]. PKA kinase as well as some PKC isozymes [12], CK2 [13] or Fyn [14] phosphorylate NRF2 modifying its stability. Previous work from our group reported that glycogen synthase kinse-3β (GSK-3β) inhibits NRF2 by nuclear exclusion and proteasomal degradation [15], [25], [26], [27], [28], [29], [30]. The molecular details will be discussed in the Section 4.1. Furthermore, NRF2 is submitted to other types of regulation. For instance, NRF2 acetylation by CBP/p300 increases its activity [17], while it is inhibited by miR153, miR27a, miR142-5p, and miR144 [16], or by methylation of cytosine-guanine (CG) islands within the NRF2 promoter [18].
Impact of Protein Aggregates on NRF2 Regulatory Mechanisms
In this section we will focus in how accumulation of misfolded protein could impact NRF2 activity providing some of the pathways mentioned above as illustrative examples. Firstly, we need to consider that protein accumulation has been tightly linked with oxidative damage. Indeed, misfolded protein accumulation and aggregation induce abnormal production of reactive oxygen species (ROS) from mitochondria and other sources [19]. As mentioned above, ROS will modify redox-sensitive cysteines of KEAP1 leading to the release, stabilization and nuclear localization of NRF2.
Regarding proteinopathies, an example of dysregulated signaling events that may affect NRF2 is provided by the hyperactivation of GSK-3β in AD. GSK-3β, also known as TAU kinase, participates in the phosphorylation of this microtubule-associated protein, resulting in its aggregation, formation of neurofibrillary tangles and interruption of axonal transport (reviewed in [20]). On the other hand, GSK-3β dramatically reduces NRF2 levels and activity as mentioned above. Although not widely accepted, the amyloid cascade proposes that toxic Aβ oligomers increase GSK-3β activity together with TAU hyper-phosphorylation and neuron death [21], [22]. There are different models to explain how Aβ favors GSK3-β activity. For instance, Aβ binds to the insulin receptor and inhibits PI3K and AKT signaling pathways, which are crucial to maintain GSK-3β inactivated by phosphorylation at its N-terminal Ser9 residue [23]. On the other hand, extracellular Aβ interacts with Frizzled receptors, blocking WNT signaling [24] and again resulting in release of active GSK-3β. In summary, Aβ accumulation leads to abnormal hyperactivation of GSK-3β, thus impairing an appropriate NRF2 response.
As discussed in the following section, misfolded proteins lead to activation of PERK and MAPKs, which in turn up-regulate NRF2 [31], [8], [9], [10], [11]. Moreover, dysregulated CBP/p300 activity has been reported in several proteinopathies [32] and a global decrease in DNA methylation in AD brains was also shown [33], therefore providing grounds to explore the relevance of these findings in NRF2 regulation.
We and others have observed in necropsies of PD and AD patients an increase in NRF2 protein levels and some of its targets, such as heme oxygenase 1 (HMOX1), NADPH quinone oxidase 1 (NQO1), p62, etc., both by immunoblot and by immunohistochemistry [34], [35], [36], [37], [38], [39]. The up-regulation of NRF2 in these diseases is interpreted as an unsuccessful attempt of the diseased brain to recover homeostatic values. However, another study indicated that NRF2 is predominantly localized in the cytoplasm of AD hippocampal neurons, suggesting reduced NRF2 transcriptional activity in the brain [40]. It is conceivable that the disparity of these observations is related to changes in the factors that control NRF2 along the progressive stages of neurodegeneration.
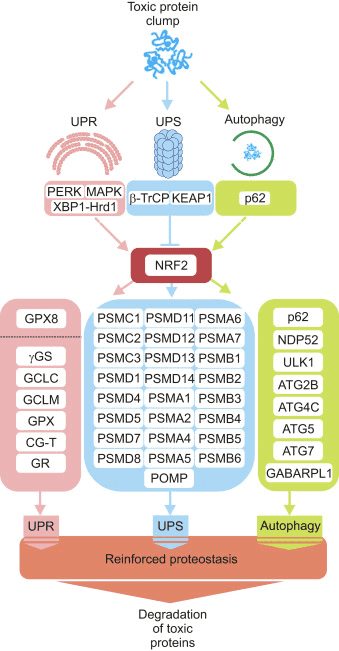
Three major systems contribute to proteostasis, namely the unfolded protein response (UPR), the ubiquitin proteasome system (UPS) and autophagy. Next, we present evidence to envision NRF2 as a hub connecting emergency signals activated by protein aggregates with the protein derivative machinery.
NRF2 Participates in the Unfolded Protein Response (UPR)
NRF2 Activation in Response to the UPR
Oxidative protein folding in the ER is driven by a number of distinct pathways, the most conserved of which involves the protein disulfide-isomerase (PDI) and the sulfhydryl oxidase endoplasmic oxidoreductin 1 (ERO1α and ERO1β in mammals) as disulfide donor. Briefly, PDI catalyzes the formation and breakage of disulfide bonds between cysteine residues within proteins, as they fold, due to the reduction and oxidation of its own cysteine aminoacids. PDI is recycled by the action of the housekeeping enzyme ERO1, which reintroduces disulfide bonds into PDI [41]. Molecular oxygen is the terminal electron acceptor of ERO1, which generates stoichiometric amounts of hydrogen peroxide for every disulfide bond produced [42]. Peroxidases (PRX4) and glutathione peroxidases (GPX7 and GPX8) are key enzymes to reduce hydrogen peroxide in the ER. When this oxido-reductive system does not work properly, abnormal accumulation of misfolded proteins occurs in the ER and a set of signals named the unfolded protein response (UPR) is transmitted to the cytoplasm and nucleus to reestablish the ER homeostasis [43]. Three membrane-associated proteins have been identified for sensing ER stress in eukaryotes: activating transcription factor 6 (ATF6), pancreatic ER eIF2α kinase (PERK, also double-stranded RNA-activated protein kinase-like ER kinase), and inositol-requiring kinase1 (IRE1). The luminal domain of each sensor is bound to a 78 kDa chaperone termed glucose-regulated protein (GRP78/BIP). BIP dissociates upon ER stress to bind unfolded proteins, leading to the activation of the three sensors [44].
NRF2 and its homologue NRF1, also related to the antioxidant response, participate in the transduction of the UPR to the nucleus. In the case of NRF1, this protein is located at the ER membrane and undergoes nuclear translocation upon deglycosylation or cleavage. Then, UPR activation leads to the processing of NRF1 and nuclear accumulation of the resulting fragment in the nuclear compartment. However, the ability to transactivate ARE-containing genes of this NRF1 fragment is still under discussion [45].
Glover-Cutter and co-workers showed activation of the NRF2 orthologue of C. elegans, SKN-1, with different ER stressors. Increased SKN-1 expression was dependent on different UPR mediators, including IRE1 or PERK worm orthologues [46]. In PERK-deficient cells, impaired protein synthesis leads to accumulation of endogenous peroxides and subsequent apoptosis [47]. The effector used by PERK to protect the ER from these peroxides might be NRF2, since it has been reported that PERK phosphorylates NRF2 at Ser40, thus preventing its degradation by KEAP1 [31]. The induction of ASK1 is also likely to play a role in this route through the TRAF2-mediated kinase action of IRE1 [48]. Although the role of MAPKs in the regulation of NRF2 is still controversial, it was recently suggested that the IRE1-TRAF2-ASK1-JNK pathway might activate NRF2 [49] (Fig. 1). Interestingly, in C. elegans and human cells, new evidence suggests that cysteine sulfenylation of IRE1 kinase at its activation loop inhibits IRE1-mediated UPR and initiates a p38 antioxidant response driven by NRF2. The data suggest that IRE1 has an ancient function as a cytoplasmic sentinel that activates p38 and NRF2 [50].
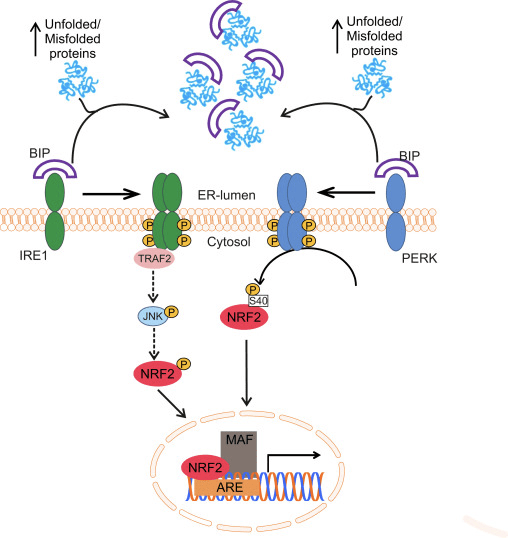
Many studies on the induction of the UPR have been conducted with the inhibitor of protein glycosylation tunicamycin. NRF2 appears to be essential for prevention of tunicamycin-induced apoptotic cell death [31] and its activation under these conditions is driven by the autophagic degradation of KEAP1 [51]. Accordingly, shRNA-mediated silencing of NRF2 expression in βTC-6 cells, a murine insulinoma β-cell line, significantly increased tunicamycin-induced cytotoxicity and led to an increase in the expression of the pro-apoptotic ER stress marker CHOP10. On the other hand, NRF2 activation by 1,2-dithiole-3-thione (D3T) reduced tunicamycin cytotoxicity and attenuated the expression of CHOP10 and PERK [52]. Interestingly, olfactory neurons submitted to systemic application of tunicamycin increased NRF2 in parallel with other UPR-members such as CHOP, BIP, XBP1 [53]. These results have been extended to in vivo studies, as lateral ventricular infusion of tunicamycin in rats induced expression of PERK and NRF2 in the hippocampus accompanied by significant cognitive deficits, increased TAU phosphorylation and Aβ42 deposits [54].
NRF2 Up-Regulates Key Genes for the Maintenance of the ER Physiology
The ER lumen needs an abundant supply of GSH from the cytosol in order to maintain disulfide chemistry. NRF2 modulates crucial enzymes of the GSH metabolism in the brain, such as cystine/glutamate transport, γ-glutamate cysteine synthetase (γ-GS), glutamate-cysteine ligase catalytic and modulator subunits (GCLC and GCLM), glutathione reductase (GR) and glutathione peroxidase (GPX) (reviewed in [55]). The relevance of NRF2 in the maintenance of GSH in the ER is supported by the finding that pharmacological or genetic activation of NRF2 results in increased GSH synthesis via GCLC/GCLM, while inhibiting the expression of these enzymes by NRF2-knockdown caused an accumulation of damaged proteins within the ER leading to the UPR activation [56].
In C. elegans several components of the UPR target genes regulated by SKN-1, including Ire1, Xbp1 and Atf6. Although NRF2 up-regulates the expression of several peroxidase (PRX) and glutathione peroxidase (GPX) genes in mammals (reviewed in [57]), only GPX8 is a bona fide ER-localized enzyme, harboring the KDEL retrieval signal [58]. Loss of GPX8 causes UPR activation, leakage of ERO1α-derived hydrogen peroxide to the cytosol and cell death. Hydrogen peroxide derived from ERO1α activity cannot diffuse from the ER to the cytosol owing to the concerted action of GPX8 and PRX4 [59]. In this regard, an analysis of the antioxidant defense pathway-gene expression array using RNA from wild type and NRF2-null mice tissue, revealed that the expression of GPX8 was down-regulated in the absence of NRF2 [60]. In line with this, transcriptome analysis from patient samples suffering from myeloproliferative neoplasms, polycythemia or myelofibrosis, diseases also associate with oxidative stress and low-grade chronic inflammation, show lower expression levels of both NRF2 and GPX8 compared with control subjects [61]. There are not yet studies that specifically involve GPX8 in human brain protection but a transcriptome analysis in mice indicates a compensatory GPX8 increase in response to the Parkinsonian toxin MPTP [62].
Impact of NRF2 on the UPR Dysregulation in Neurodegenerative Diseases
Malfunction of PDI enzymes and chronic activation of the UPR might subsequently initiate or accelerate neurodegeneration. Disease-affected neurons, animal models of neurodegenerative disease as well as post-mortem human tissues evidenced up-regulation of several UPR-markers in most of these disorders. The alteration of PDI/UPR pathway in neurodegenerative diseases has been nicely reviewed in [63] but the following highlights from brain post-mortem samples should be considered. PDI levels are increased in tangle-bearing neurons and in Lewy Bodies of AD and PD patients, respectively [64], [65]. PDI and ERP57 are up-regulated in CSF from ALS patients and in brains from CJD subjects [66], [67], [68]. BIP, PERK, IRE1 and ATF6 are elevated in samples from patients with AD, PD or ALS [69], [70], [71], [67]. BIP, CHOP and XBP1 are elevated in post-mortem brain samples from HD [72], [73]. Moreover, up-regulation of ERP57, GRP94 and BIP was found in cortex tissues from CJD patients [74]. Altogether, this evidence reveals that the accumulation of misfolded proteins in the brain parenchyma leads to a deleterious and chronic activation of the UPR. Interestingly, there is a recent study linking activation of NRF2 by PERK in early AD. In this study, the authors analyzed whether oxidative stress mediated changes in NRF2 and the UPR may constitute early events in AD pathogenesis by using human peripheral blood cells and an AD transgenic mouse model at different disease stages. Increased oxidative stress and increased pSer40-NRF2 were observed in human peripheral blood mononuclear cells isolated from individuals with mild cognitive impairment. Moreover, they reported impaired ER calcium homeostasis and up-regulated ER-stress markers in these cells from individuals with mild cognitive impairment and mild AD [75].
Mutual Regulation of NRF2 and the Ubiquitin Proteasome System (UPS)
The UPS Modulates NRF2 Protein Levels
The UPS participates in the degradation of damaged or misfolded proteins and controls the levels of key regulatory molecules in the cytosol and the nucleus. The central core of this system is a large multisubunit enzyme that contains a proteolytic active complex named 20S. The 20S core proteasome degrades unfolded proteins, but binding to different regulatory protein complexes changes its substrate specificity and activity. For instance, the addition of one or two 19S regulatory subunits to the 20S core constitutes the 26S proteasome and changes its specificity towards native folded proteins [76], [77]. Proteasomal degradation needs covalent binding of ubiquitin. Conjugation of ubiquitin proceeds via a three-step cascade mechanism. First, the ubiquitin-activating enzyme E1 activates ubiquitin in an ATP-requiring reaction. Then, one E2 enzyme (ubiquitin-carrier protein or ubiquitin-conjugating enzyme) transfers the activated ubiquitin from E1 to the substrate that is specifically bound to a member of the ubiquitin-protein ligase family, named E3. Although the exact fate of the ubiquitinated-protein will depend on the nature of the ubiquitin chain, this process generally results in the degradation by the 26S proteasome [78].
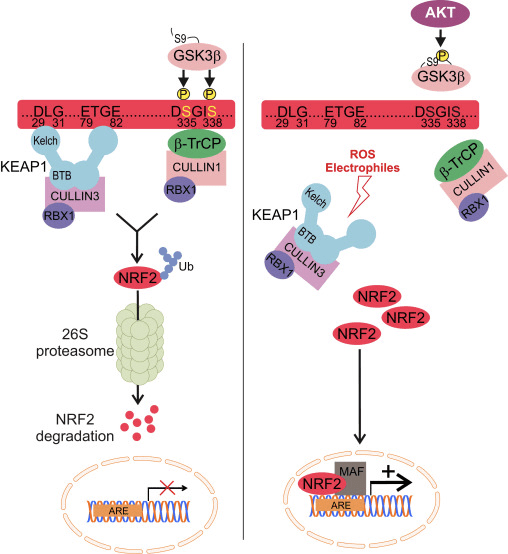
The E3-ligase KEAP1 is the best known inhibitor of NRF2. The mechanism of KEAP1 regulation elegantly explains how NRF2 levels adjust to oxidant fluctuations. Under basal conditions, newly synthesized NRF2 is grabbed by the homodimer KEAP1, which binds one NRF2 molecule at two amino acid sequences with low (aspartate, leucine, glycine; DLG) and high (glutamate, threonine, glycine, glutamate; ETGE) affinity. The interaction with KEAP1 aids to present NRF2 to the CULLIN3/RBX1 protein complex, resulting in its ubiquitination and subsequent proteasomal degradation. However, redox modification of KEAP1 impedes presentation of NRF2 to the UPS represented by CULLIN3/RBX1. As a result, newly synthetized NRF2 escapes KEAP1-dependent degradation, accumulates in the nucleus and activates ARE-containing genes [79], [80], [81], [82].
The E3-ligase adaptor β-TrCP is also a homodimer that participates in the signaling events related to the phosphorylation of NRF2 by GSK-3β. This kinase phosphorylates specific serine residues of NRF2 (aspartate, serine, glycine, isoleucine serine; DSGIS) to create a degradation domain that is then recognized by β-TrCP and tagged for proteasome degradation by a CULLIN1/RBX1 complex. The identification of the specific amino acids that are phosphorylated by GSK-3β in this degron was conducted by a combination of site-directed mutagenesis of the Neh6 domain, 2D-gel electrophoresis [15], [26] and mass spectroscopy [83]. Consequently, inhibition of GSK-3β by highly selective drugs or siRNAs against GSK-3 isoforms resulted in an increase in NRF2 protein levels. Similar results were found with siRNAs against β-TrCP isoforms 1 and 2. Stabilization of NRF2 following GSK-3β inhibition occurred in KEAP1-deficient mouse embryo fibroblasts and in an ectopically expressed NRF2 deletion mutant lacking the critical ETGE residues for high-affinity binding to KEAP1, further demonstrating a KEAP1-independent regulation.
In the context of neurodegenerative diseases, we can envision the modulation of NRF2 by the UPS in two different ways. On the one hand, the KEAP1 system would sense redox imbalance derived from misfolded protein accumulation, while GSK-3/β-TrCP axis would act as an active participant in signaling transduction altered by loss of proteostasis (Fig. 2).
NRF2 Increases UPS Activity Through the Transcriptional Control of Proteasome Subunits
NRF2 up-regulates the expression of several proteasome subunits, thus protecting the cell from the accumulation of toxic proteins. Twenty proteasome- and ubiquitination-related genes appear to be regulated by NRF2, according to a wide microarray analysis from liver RNA that was set up with the NRF2 inducer D3T [84]. In a posterior study, the same authors evidenced that the expression of most subunits of the 26S proteasome were enhanced up to three-fold in livers from mice treated with D3T. Subunit protein levels and proteasome activity were coordinately increased. However, no induction was seen in mice where the transcription factor NRF2 was disrupted. Promoter activity of the PSMB5 (20S) proteasome subunit increased with either NRF2 overexpression or treatment with activators in mouse embryonic fibroblasts, and AREs were identified in the proximal promoter of PSMB5 [85]. Pharmacological activation of NRF2 resulted in elevated expression levels of representative proteasome subunits (PSMA3, PSMA6, PSMB1 and PSMB5) only in non-senescent human fibroblasts containing functional NRF2 [86]. NRF2 activation during adaptation to oxidative stress results in high expression of the PSMB1 (20S) and PA28α subunits (or S11, proteasome regulator) [87]. Moreover, results from human embryonic stem cells revealed that NRF2 controls the expression of the proteasome maturation protein (POMP), a proteasome chaperone, which in turn modulates the proliferation of self-renewing human embryonic stem cells, three germ layer differentiation and cellular reprogramming [88]. All together, these studies indicate that NRF2 up-regulates the expression of key components of the UPS and therefore actively contributes to the clearance of proteins that otherwise would be toxic.
The NRF2-UPS Axis in Neurodegenerative Diseases
The role of the UPS in neurodegenerative diseases is a field of intensive debate. Initial studies reported decreased proteasome activity in human necropsies of patients affected from several neurodegenerative diseases. However, other studies employing in vitro and in vivo approaches found unchanged or even increased proteasome activity (reviewed in [89]). One possible explanation for this discrepancy is that the levels of the UPS components might change during disease progression and in different brain regions as is has been suggested for NRF2-targets.
Despite this controversy, it should be noted that up-regulation of ARE-containing proteasome genes will reinforce the UPS by increasing the clearance of toxic proteins in the brain. Indeed, ablation of NRF1, also modulator of the antioxidant response, in neuronal cells leads to impaired proteasome activity and neurodegeneration. Chromatin immunoprecipitation experiments and transcriptional analysis demonstrated that PSMB6 is regulated by NRF1. In addition, gene expression profiling led to the identification of NRF1 as a key transcriptional regulator of proteasome genes in neurons, suggesting that perturbations in NRF1 may contribute to the pathogenesis of neurodegenerative diseases [90]. Interestingly, NRF1 and its long isoform called TCF11 were shown to up-regulate ARE-containing proteasome genes upon proteasome inhibition in a feedback loop to compensate for reduced proteolytic activity [91], [92].
Regarding NRF2, there is a correlation among reduction of NRF2, RPT6 (19 S) and PSMB5 (20 S) levels in the midbrain of DJ-1-deficient mice treated with the neurotoxin paraquat [93]. Moreover, the naturally-occurring compound sulforaphane (SFN) gives a more robust image of NRF2 as a crucial modulator of the UPS. In vitro experiments with murine neuroblastoma Neuro2A cells evidenced an enhanced expression of the catalytic subunits of the proteasome, as well as its peptidase activities in response to SFN. This drug protected cells from hydrogen peroxide-mediated cytotoxicity and protein oxidation in a manner dependent on proteasome function [94]. In addition, Liu and co-workers employed a reporter mouse to monitor the UPS activity in response to SFN in the brain. These mice ubiquitously express the green fluorescence protein (GFP) fused to a constitutive degradation signal that promotes its rapid degradation by the UPS (GFPu). In cerebral cortex, SFN reduced the level of GFPu with a parallel increase in chymotrypsin-like (PSMB5), caspase-like (PSMB2), and trypsin-like (PSMB1) activities of the 20 S proteasome. In addition, treatment of Huntington-derived cells with SFN revealed that NRF2 activation enhanced mHtt degradation and reduced mHtt cytotoxicity [95]. The major mechanism of SFN action is through induction of NRF2 [96]. The specific contribution of NRF2 should be addressed employing NRF2-null systems in further studies.
Functional Connection Between NRF2 and Macroautophagy
NRF2 Protein Levels are Modulated by the Adaptor Protein P62
Autophagy refers to the degradation of cytosolic components inside lysosomes. This process is used for the clearance of long-lived and misfolded proteins as well as damaged organelles. A direct link between NRF2 and autophagy was first observed in connection with the adaptor protein p62, also termed SQSTM1 [97], [98], [99], [100], [101]. This protein shuttles ubiquitinated proteins to the proteasomal and lysosomal degradation machineries and sequesters damaged proteins into aggregates prior to their degradation. P62 presents an ubiquitin-associated (UBA) domain, for binding to ubiquitinated proteins, and a LC3-interacting region (LIR) for integration with the autophagosomal membrane through the autophagy receptor LC3.
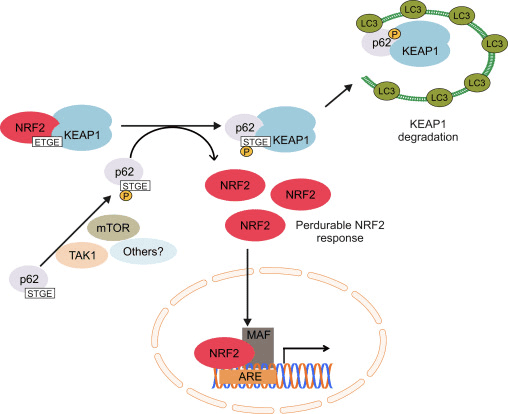
Although the p62-mediated induction of NRF2 and its target genes was first reported in 2007 [102], the molecular mechanism was not fully understood until the discovery of its interaction with KEAP1 [103], [98], [99], [100], [101]. Komatsu and coworkers identified a KEAP1 interacting region (KIR) in p62 that bound KEAP1 in the same basic surface pocket as NRF2 and with a binding affinity similar to the ETGE motif in NRF2, suggesting competition between p62 and NRF2. The phosphorylation of Ser351 in the KIR motif in p62 (349-DPSTGE-354) was shown to increase its affinity for KEAP1, competing with NRF2 binding and allowing its accumulation and transcriptional activation of its target genes [98], [99]. In fact, p62 overexpression led to reduced NRF2 ubiquitination and consequent stabilization as well as induction of its target genes [104]. Some kinases have been suggested to participate in p62 phosphorylation. The mammalian target of rapamycin complex 1 (mTORC1) may be implicated, as treatment with the mTOR inhibitor rapamycin suppressed the phosphorylation of p62 and the down-regulation of KEAP1 upon arsenite treatment. Recently, it was demonstrated that TGF-β-activated kinase 1 (TAK1) could also phosphorylate p62, enhancing KEAP1 degradation and NRF2-up-regulation. The authors of this study suggest this is a way to regulate cellular redoxtasis under steady-state conditions, as TAK1-deficiency up-regulates ROS in the absence of any exogenous oxidant in different mouse tissues in parallel with a reduction in NRF2 protein levels [105].
A p62 construct lacking the UBA domain was still capable of binding KEAP1, implying that the interaction did not depend on ubiquitinated KEAP1 [101]. However, the p62 homologue in Drosophila melanogaster, named Ref(2), does not contain a KIR motif and does not directly interact with DmKEAP1, although it can bind to ubiquitinated DmKEAP1 through the UBA domain. Moreover, DmKEAP1 can directly interact with Atg8 (homologue to mammalian LC3). KEAP1 deficiency results in Atg8 and autophagy induction dependent on the NRF2 orthologue CncC and independent on TFEB/MITF [106]. The relationship between NRF2 and autophagy seems to be conserved though, highlighting its functional relevance.
The induction of NRF2 by p62 is the result of both the competition to bind KEAP1 and degradation of KEAP1 in the lysosome. Silencing of p62 with siRNA doubled KEAP1 half-life in parallel with a decrease in NRF2 and its target genes [101]. In agreement, ablation of p62 expression evidenced increased levels of KEAP1 compared with wild type mice. Very relevant, the increment in KEAP1 levels was not affected by proteasome inhibitors but was reduced under starvation-inducing autophagy [107]. In fact, KEAP1 is present in mammalian cells in autophagic vesicles decorated with p62 and LC3 [99], [100], [103]. All these data suggest that KEAP1 is a substrate of the macroautophagy machinery, but this issue should be analyzed with more detail because of the existence of some controversial results. KEAP1 protein levels were increased in Atg7-null mice, a key effector of macroautophagy [107], but pharmacological inhibition of macroautophagy with torin1, E64/pepstatin or bafilomycin failed to accumulate KEAP1 [107], [100]. Overall, these results suggest that increased p62 levels sequester KEAP1 into autophagic vacuoles and probably these results in KEAP1 autophagic degradation allowing NRF2 activation (Fig. 3). Two different studies reported that the sulfinic acid reductases SESTRINS play an important role in this context. SESTRIN 2 interacts with p62, KEAP1 and RBX1 and facilitates p62-dependent degradation of KEAP1 and NRF2 activation of target genes [108]. Another study showed that SESTRIN 2 interacted with ULK1 and p62, promoting phosphorylation of p62 at Ser403 which facilitated degradation of cargo proteins including KEAP1 [109].
Modulation of Macroautophagy Genes by NRF2
NRF2 regulates the expression of relevant genes for macroautophagy as well as it does for the UPR and the UPS. The first evidence came from studies in which p62 expression was shown to be induced upon exposure to electrophiles, ROS and nitric oxide [110], [111], [112]. The mechanism of induction was described some years later with the finding that p62 contains a functional ARE in its gene promoter [99]. In a recent study, several other functional AREs were found and validated following bioinformatics analysis and ChIP assays. Moreover, mouse embryonic fibroblasts and cortical neurons from Nrf2-knockout mice exhibited reduced p62 expression, which could be rescued with an NRF2-expressing lentivirus. Similarly, NRF2 deficiency reduced p62 levels in injured neurons from mice hippocampus [36]. Therefore, it has been suggested that NRF2 activation increases p62 levels, resulting in KEAP1 degradation and favoring further NRF2 stabilization in a positive feedback loop. This non-canonical mechanism of NRF2 induction requires changes in gene expression and might be a relevant response to prolonged cellular stress.
The cargo recognition protein NDP52 was shown to be transcriptionally regulated by NRF2. NDP52 works in a similar way to p62, recognizing ubiquitinated proteins and interacting with LC3 through a LIR domain, so that cargoes are degraded in lysosomes. Five putative AREs were found in Ndp52 promoter DNA sequence. Three of them were identified with different mutant constructs and ChIP assays as indispensable for NRF2-mediated Ndp52 transcription [113]. Of note, Ndp52 mRNA levels were reduced in the hippocampus of Nrf2-knockout mice. One of these sequences was also validated in an independent study as an NRF2-regulated ARE [36].
However, the role of NRF2 in the modulation of autophagy is not limited to the induction of these two cargo-recognition proteins. In order to gain deeper insight in the role of NRF2 in the modulation of additional autophagy-related genes, our group screened the chromatin immunoprecipitation database ENCODE for two proteins, MAFK and BACH1, which bind the NRF2-regulated AREs. Using a script generated from the JASPAR’s consensus ARE sequence, we identified several putative AREs in many autophagy genes. Twelve of these sequences were validated as NRF2 regulated AREs in nine autophagy genes, whose expression was diminished in mouse embryo fibroblasts of Nrf2-knockout mice but could be restored by an NRF2-expressing lentivirus. Our study demonstrated that NRF2 activates the expression of some genes involved in different steps of the autophagic process, including autophagy initiation (ULK1), cargo recognition (p62 and NDP52), autophagosome formation (ATG4D, ATG7 and GABARAPL1), elongation (ATG2B and ATG5), and autolysosome clearance (ATG4D). Consequently, autophagy flux in response to hydrogen peroxide was impaired when NRF2 was absent [36].
Relevance of NRF2-Mediated Macroautophagy Genes Expression in Neurodegenerative Disorders
Defective autophagy has been shown to play an important role in several neurodegenerative diseases [114] and ablation of autophagy leads to neurodegeneration in mice [115], [116]. Atg7-knockout mice revealed that autophagy deficiency results in p62 accumulation in ubiquitin-positive inclusion bodies. KEAP1 was sequestered in these inclusion bodies, leading to NRF2 stabilization and induction of target genes [103]. Importantly, excessive accumulation of p62 together with ubiquitinated proteins has been identified in neurodegenerative diseases, including AD, PD and ALS [117]. In fact, neurons expressing high levels of APP or TAU of AD patients also expressed p62 and nuclear NRF2, suggesting their attempt to degrade intraneuronal aggregates through autophagy [36].
NRF2 deficiency aggravates protein aggregation in the context of AD. In fact, increased levels of phosphorylated and sarkosyl-insoluble TAU are found in Nrf2-knockout mice, although no difference in kinase or phosphatase activities could be detected comparing with the wild-type background [113]. Importantly, NDP52 was demonstrated to co-localize with TAU in murine neurons and direct interaction between phospho-TAU and NDP52 was shown by co-immunoprecipitation experiments both in mice and AD samples, pointing to its role in TAU degradation. Interestingly, silencing of NDP52, p62 or NRF2 in neurons resulted in increased phospho-TAU [113], [118]. Moreover, increased intraneuronal APP aggregates were found in the hippocampus of APP/PS1ΔE9 mice when NRF2 was absent. This correlated with altered autophagy markers, including increased phospho-mTOR/mTOR and phospho-p70S6k/p70S6k ratios (indicative of autophagy inhibition), augmented levels of pre-cathepsin D and a larger number of multivesicular bodies [119]. In mice co-expressing human APP (V717I) and TAU (P301L), NRF2 deficiency led to increased levels of total and phospho-TAU in the insoluble fraction and increased intraneuronal APP aggregates, together with reduced neuronal levels of p62, NDP52, ULK1, ATG5 and GABARAPL1. Co-localization between the adaptor protein p62 and APP or TAU was reduced in the absence of NRF2 [36]. Overall, these results highlight the importance of NRF2 in neuronal autophagy.
Different Transcription Factors Act Coordinately to Modulate Proteostasis
Under steady state conditions, proteostasis is controlled via protein-protein interactions and post-translational modifications obtaining a rapid response. However, cellular adaptation requires the transcriptional regulation of the UPR, UPS and autophagy genes. Considering that nerve cells are continuously submitted to low-grade toxic insults, including oxidative and proteotoxic stress, a reinforcement of proteostasis induced by transcriptional modulation might help preventing brain degeneration.
In the case of the UPR, the activation of each of the three arms will finally result in the transcriptional induction of certain genes (reviewed in [43]). For instance, an ATF6-derived fragment (ATF6f) binds to ER-stress response elements (ERSE) and induces the expression of several genes, including XBPI, BIP and CHOP. In addition, PERK signaling leads to the activation of the transcription factor ATF4, which controls the expression of multiple UPR-related genes and some others including the NRF2 target genes Hmox1 and p62. Finally, IRE1 activation results in the generation of an active transcription factor, spliced XBP1 (XBP1s), which controls the transcription of genes encoding proteins involved in protein folding.
On the other hand, NRF1 was shown to be necessary for proteasomal gene expression in the brain, as Nrf1-knockout mice exhibited reduced expression of genes encoding various subunits of the 20S core, as well the 19S regulatory complex together with impaired proteasomal function [90]. Both NRF1 and NRF2 bind to ARE sequences in the promoter regions of its target genes, which suggests they have overlapping transcriptional activities, although they differ in their regulatory mechanisms and cellular localization [120].
Transcription factors of the Forkhead box O (FOXO) family control the expression of multiple autophagy-related genes. Similar to what occurs with NRF2, there are multiple layers of regulation of the activity of FOXO members, which can be induced upon nutritional or oxidative stress [121]. Finally, the transcription factor TFEB, considered the master regulator of lysosomal biogenesis, plays a crucial role in regulation of autophagy under nutritional stress conditions. Thus, inhibition of mTORC1 leads to nuclear translocation of TFEB and induction of the expression of autophagy genes [122].
Overall, the existence of different transcriptional regulators of these machineries also suggests crosstalk and partially redundant mechanisms that may assure proteostasis under different circumstances. Accordingly, NRF2 may have a relevant role in tissues that support high levels of oxidative stress. For instance, oxidative stress-induced NRF2 may function under nutrient-rich conditions to transcriptionally up-regulate autophagy, similar to what has been found for TFEB under starvation conditions. Moreover, the brain functions largely under nutrient-rich conditions, posing NRF2 as a relevant mechanism to activate autophagy in neurons.
Promising Therapeutic Potential for NRF2 in Proteinopathies
In the past few years, a great progress has been made in the knowledge of the regulatory roles of the UPR, UPS
However, a key remaining question is whether it will be useful or deleterious to increase NRF2 levels in brain. Analysis of epidemiological data may provide a partial answer, as it indicates that the NFE2L2 gene is highly polymorphic and some single nucleotide polymorphisms found in its promoter regulatory region may provide a range of “physiological†variability in gene expression at the population level and some haplotypes were associated with decreased risk and/or delayed onset of AD, PD or ALS [123]. Moreover, as discussed by Hayes and colleagues [124], NRF2 effect might have an U-shaped response, meaning that too low NRF2 levels may result in a loss of cytoprotection and increased susceptibility to stressors, while too much NRF2 might disturb homeostatic balance towards a reductive scenario (reductive stress), which would favor protein misfolding and aggregation. Low NRF2 levels in the brain support the idea that a slight up-regulation may be sufficient to achieve a benefit under pathological conditions. In fact, the protective role of pharmacological NRF2-mediated activation of protein clearance has been shown in different neurodegeneration cell culture and in vivo models.
SFN is a pharmacological NRF2 activator that was demonstrated to induce proteasomal and autophagy gene expression [95], [36]. Interestingly, Jo and colleagues demonstrated that SFN reduced the levels of phosphorylated TAU and increased Beclin-1 and LC3-II, suggesting NRF2 activation may facilitate degradation of this toxic protein through autophagy [113]. Moreover, degradation of mHtt was enhanced with SFN, and this was reverted with the use of MG132, indicating proteasomal degradation of this toxic protein [95]. Autophagy-mediated degradation of phospho- and insoluble-TAU was reported with the organic flavonoid fisetin. This compound was able to induce autophagy by simultaneously promoting the activation and nuclear translocation of both TFEB and NRF2, along with some of its target genes. This response was prevented by TFEB or NRF2 silencing [125]. Bott and colleagues reported beneficial effects of a simultaneous NRF2, NRF1 and HSF1 activator on protein toxicity in spinal and bulbar muscular atrophy, a neurodegenerative disorder caused by expansion of polyglutamine-encoding CAG repeats in which protein aggregates are present [126]. The potential of NRF2 activation for the treatment of neurodegenerative disorders has been demonstrated with the approval of BG-12, the oral formulation of the NRF2 inducer dimethyl fumarate (DMF), for the treatment of multiple sclerosis [127], [128]. The success of DMF with autoimmune diseases with a strong inflammatory component suggests that neurodegenerative diseases might benefit from repositioning this drug. In a recent preclinical study of an α-synucleinopathy model of PD, DMF was shown to be neuroprotective due, in part, to its induction of autophagy [129]. Studies reporting beneficial effects of NRF2 on neurodegeneration but not focusing on its effect on protein clearance are even more abundant (for a comprehensive review, see [7]). This is quite relevant, as it highlights the multiple damaging processes that can be simultaneously targeted by a single hit in NRF2, also including oxidative stress, neuroinflammation or mitochondrial dysfunction. However, future work will be needed to definitely determine if pharmacological activation of NRF2 may be a valid strategy to facilitate degradation of toxic proteins in the brain.
As explained before, exacerbated GSK-3β activity was reported in neurodegenerative diseases and it has been speculated that consequent NRF2 reduction can be partially responsible for the deleterious outcome. Under these pathological conditions, GSK-3 inhibitors could also cooperate to increase NRF2 levels and proteostasis. The beneficial effects of GSK-3 inhibitors have been reported in different models of neurodegeneration and, more interesting, GSK-3 repression was shown to reduce the levels of toxic proteins [130], [131], [132], [133]. Although no direct links between GSK-3 inhibition and NRF2-transcriptional regulation of genes promoting proteostasis have been observed yet, it is reasonable to speculate that down-regulation of GSK-3 activity would result in increased NRF2 levels, which eventually will result in reinforced proteostasis.
The transcriptional activity of NRF2 as well as the cellular capacity to maintain proteostasis decrease with age, the main risk factor for the development of neurodegenerative diseases. It is reasonable to think that the reinforcement of NRF2 and, consequently, proteostasis would, at least, delay the accumulation of protein aggregates and neurodegeneration. Indeed, treatment of human senescent fibroblasts with 18α-glycyrrhetinic acid (18α-GA) triterpenoid promoted NRF2 activation, leading to proteasome induction and enhanced life span. This study suggests that pharmacological activation of NRF2 is possible even in late life [86]. Moreover, a later study showed that this compound mediated SKN-1 and proteasome activation in C.elegans with beneficial effects on AD progression in relevant nematode models [134].
All things considered, NRF2-mediated induction of proteostasis-related genes seems to be beneficial in different proteinopathies.
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this
Key sections:
- 00:01:14 – Cancer and mortality
- 00:19:04 – Aging
- 00:26:30 – Brain and behavior
- 00:38:06 – Final recap
- 00:40:27 – Dose
Full timeline:
- 00:00:34 – Introduction of sulforaphane, a major focus of the video.
- 00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
- 00:02:12 – Prostate cancer risk.
- 00:02:23 – Bladder cancer risk.
- 00:02:34 – Lung cancer in smokers risk.
- 00:02:48 – Breast cancer risk.
- 00:03:13 – Hypothetical: what if you already have cancer? (interventional)
- 00:03:35 – Plausible mechanism driving
the cancer and mortality associative data. - 00:04:38 – Sulforaphane and cancer.
- 00:05:32 – Animal evidence showing
strong effect of broccoli sprout extract on bladder tumor development in rats. - 00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
- 00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
- 00:08:32 – Inhibition of breast cancer stem cells.
- 00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
- 00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
- 00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
- 00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
- 00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
- 00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
- 00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
- 00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
- 00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
- 00:19:04 – Beginning of
aging section. - 00:19:21 – Sulforaphane-enriched diet enhances
lifespan of beetles from 15 to 30% (in certain conditions). - 00:20:34 – Importance of low inflammation for longevity.
- 00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
- 00:23:40 – Mid-video recap: cancer, aging sections
- 00:24:14 – Mouse studies suggest sulforaphane might improve adaptive immune function in old age.
- 00:25:18 – Sulforaphane improved hair growth in a mouse model of balding.
Picture at 00:26:10. - 00:26:30 – Beginning of brain and behavior section.
- 00:27:18 – Effect of broccoli sprout extract on autism.
- 00:27:48 – Effect of glucoraphanin on schizophrenia.
- 00:28:17 – Start of depression discussion (plausible mechanism and studies).
- 00:31:21 – Mouse study using 10 different models of stress-induced depression show sulforaphane similarly effective as fluoxetine (
prozac ). - 00:32:00 – Study shows direct ingestion of glucoraphanin in mice is similarly effective at preventing depression from social defeat stress model.
- 00:33:01 – Beginning of neurodegeneration section.
- 00:33:30 – Sulforaphane and Alzheimer’s disease.
- 00:33:44 – Sulforaphane and Parkinson’s disease.
- 00:33:51 – Sulforaphane and Hungtington’s disease.
- 00:34:13 – Sulforaphane increases heat shock proteins.
- 00:34:43 – Beginning of traumatic brain injury section.
- 00:35:01 – Sulforaphane injected immediately after TBI improves memory (mouse study).
- 00:35:55 – Sulforaphane and neuronal plasticity.
- 00:36:32 – Sulforaphane improves learning in
model of type II diabetes in mice. - 00:37:19 – Sulforaphane and
duchenne muscular dystrophy. - 00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
- 00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
- 00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
- 00:41:01 – Anecdotes on sprouting at home.
- 00:43:14 – On cooking temperatures and sulforaphane activity.
- 00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
- 00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
- 00:44:56 – Cooking techniques and cruciferous vegetables.
- 00:46:06 – Isothiocyanates as goitrogens.

The nuclear factor erythroid-derived 2 (NF-E2)-related factor 2, otherwise known as Nrf2, is a transcription factor which regulates the expression of a variety of antioxidant and detoxifying enzymes. Research studies have also demonstrated its role in controlling oxidative stress. Most neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, are characterized by oxidative stress and chronic inflammation, the common targets of Nrf2 treatment approaches.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Concluding Remarks
Transcription factor NRF2 orchestrates a
Acknowledgements
Sciencedirect.com/science/article/pii/S2213231716304050
According to the article above, while the symptoms of neurodegenerative diseases can be treated through a variety of treatment options, research studies have demonstrated that Nrf2 activation can be a helpful treatment approach. Because Nrf2 activators target broad mechanisms of disease, all neurodegenerative diseases can benefit from the use of the Nrf2 transcription factor. The findings of Nrf2 have revolutionized the treatment of neurodegenerative diseases. The scope of our information is limited to chiropractic and spinal health issues. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at
Curated by Dr. Alex Jimenez
Referenced from: Sciencedirect.com

Additional Topic Discussion: Relieving Knee Pain without Surgery
Knee pain is a well-known symptom which can occur due to a variety of knee injuries and/or conditions, including sports injuries. The knee is one of the most complex joints in the human body as it is made-up of the intersection of four bones, four ligaments, various tendons, two menisci, and cartilage. According to the American Academy of Family Physicians, the most common causes of knee pain include patellar subluxation, patellar tendinitis or jumper’s knee, and Osgood-Schlatter disease. Although knee pain is most likely to occur in people over 60 years old, knee pain can also occur in children and adolescents. Knee pain can be treated at home following the RICE methods, however, severe knee injuries may require immediate medical attention, including chiropractic care.

EXTRA EXTRA | IMPORTANT TOPIC: Recommended El Paso, TX Chiropractor
***
Post Disclaimers
General Disclaimer, Licenses and Board Certifications *
Professional Scope of Practice *
The information herein on "Understanding Nrf2 and its Impact on Neurodegenerative Diseases" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and on our family practice-based chiromed.com site, focusing on naturally restoring health for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine; wellness; contributing etiological viscerosomatic disturbances within clinical presentations; associated somato-visceral reflex clinical dynamics; subluxation complexes; sensitive health issues; and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and licensure jurisdiction. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that directly or indirectly relate to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933





Comments are closed.