

Thyroid & Autoimmune Disease:
Key Words:
- Autoimmune thyroid disease
- Smoking
- Environmental factors
- Endogenous factors
- Accelerator hypothesis
- Selenium intake
- Iodine intake
Abstract
The thyroid gland plays a major role in the human body; it produces the hormones necessary for appropriate energy levels and an active life. These hormones have a critical impact on early brain development and somatic growth. At the same time, the thyroid is highly vulnerable to autoimmune thyroid diseases (AITDs). They arise due to the complex inter- play of genetic, environmental, and endogenous factors, and the specific combination is required to initiate thyroid autoimmunity. When the thyroid cell becomes the target of autoimmunity, it interacts with the immune system and appears to affect disease progression. It can produce different growth factors, adhesion molecules, and a large array of cytokines. Preventable environmental factors, including high iodine intake, selenium deficiency, and pollutants such as tobacco smoke, as well as infectious diseases and certain drugs, have been implicated in the development of AITDs in genetically predisposed individuals. The susceptibility of the thyroid to AITDs may come from the complexity of hormonal synthesis, peculiar oligoelement requirements, and specific capabilities of the thyroid cell’s defense system. An improved understanding of this interplay could yield novel treatment pathways, some of which might be as simple as identifying the need to avoid smoking or to control the in- take of some nutrients.
Introduction
The thyroid gland is important in the human body because of its ability to produce hormones necessary for appropriate energy levels and an active life. These molecules have pleiotropic effects, playing critical roles in early brain development, somatic growth, bone maturation, and the mRNA synthesis of more than 100 proteins that constantly regulate each and every bodily function.
At the same time, the thyroid is highly vulnerable to autoimmune diseases. The incidence of chronic autoimmune thyroiditis (CAT) and Graves’ disease (GD) has in- creased dramatically over the past few decades, afflicting up to 5% of the general population. In children, CAT is the most common cause of acquired hypothyroidism in non-endemic goiter areas.
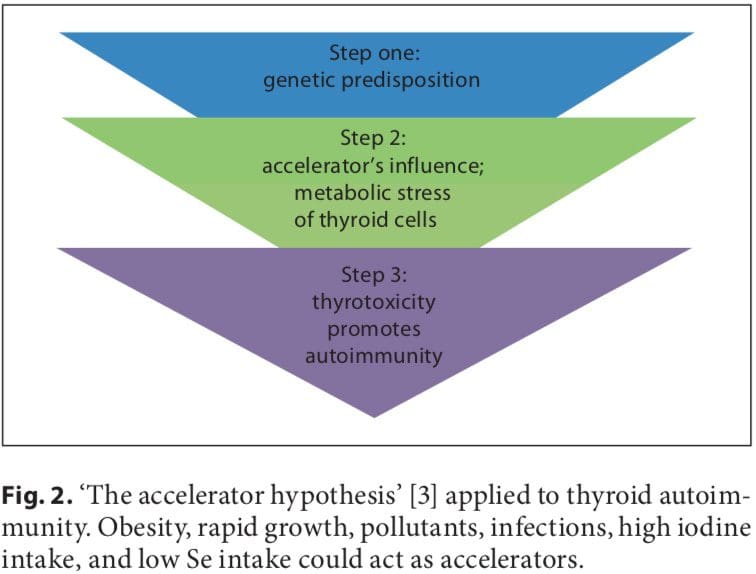
Initial studies on the association between early fetal nutrition and the pathogenesis of autoimmune thyroid diseases (AITDs) resulted in controversial data. In twin studies, Phillips et al. [1] found that among monozygotic twins the smaller twin had higher levels of thyroid per- oxidase (TPO) antibodies. However, these data were not confirmed in another twin study in which a larger cohort was analyzed [2]. The ‘accelerator hypothesis’ and the influence of rapid childhood growth due to energy-dense food and adipokine imbalance have not been investigated in childhood AITDs. In both type 1 and type 2 diabetes, the accelerator hypothesis proposes a critical influence of obesity as an exogenous factor contributing to disease; even in a population of children with type 1 diabetes, the fattest presented with disease the earliest (evidence of true acceleration) [3]. With regard to AITDs, other accelerators in addition to obesity include low selenium (Se) and a high iodine intake. Obese children are hyperleptinemic, and leptin, with its numerous functions including the promotion of cell-mediated immune responses, is a good candidate for contributing to the pathogenesis of autoimmune diseases. Obese children have been found to have increased interferon (IFN)- -secreting T helper cells and altered thyroid structure and hormonal status [4–8].
Autoimmunity is generally considered to be only a cause of disease; nevertheless, human T cell repertoires naturally comprise autoimmune lymphocytes. Autoimmune T cells can help heal damaged tissues, indicating that natural autoimmunity can also contribute to health and benefit self-maintenance [9]. The immune system makes its decisions and acts by integrating multiple signals in an ongoing dialog with tissues. It is likely that the tissue itself provides signals that trigger the type of inflammation that is required for tissue self-maintenance and repair [9, 10].
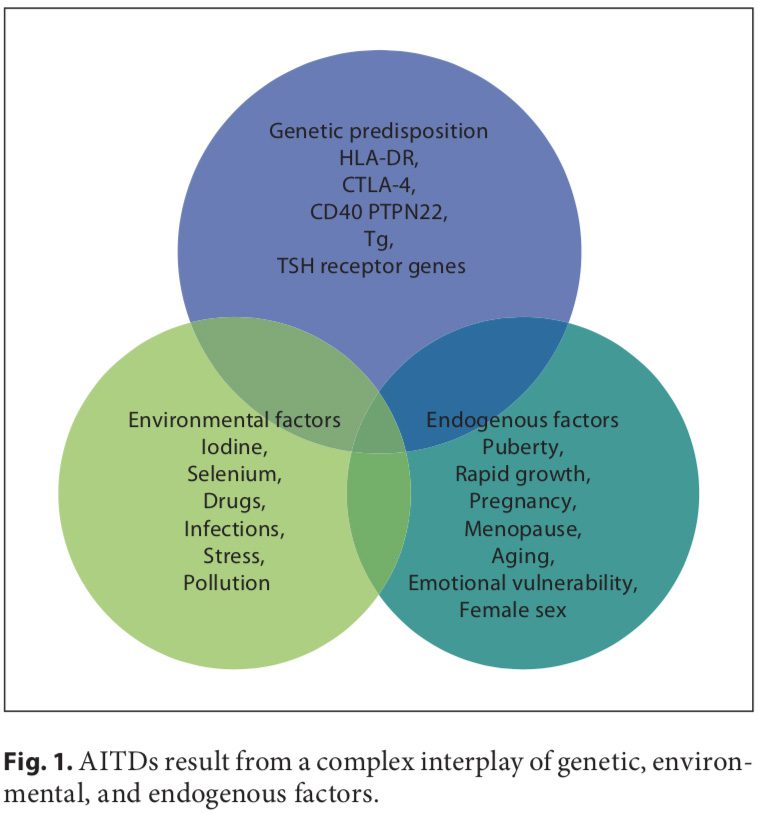 Autoimmune disorders result from a complex interplay of genetic, environmental, and endogenous factors (fig. 1), and a combination of these factors is required to initiate thyroid autoimmunity [11, 12]. Recent advances in genome-wide studies have made it possible to efficient- ly identify complex disease-associated genes. Using both the candidate gene approach and whole-genome linkage studies, 6 AITD susceptibility genes have been identified and confirmed; the first group includes the immunomodulatory gene products HLA-DR, CD40, cytotoxic T lymphocyte-associated factor (CTLA-4), and protein tyrosine phosphatase 22 (PTPN22), and the second group includes the thyroid-specific gene products thyroglobulin (Tg) and thyroid-stimulating hormone receptor (TSHR). Genetic factors predominate, accounting for approximately 80% of the likelihood of developing AITDs, whereas at least 20% is due to environmental factors (fig. 1). In recent years, a number of excellent reviews have been published on the genetic background of AITDs [13, 14].
Autoimmune disorders result from a complex interplay of genetic, environmental, and endogenous factors (fig. 1), and a combination of these factors is required to initiate thyroid autoimmunity [11, 12]. Recent advances in genome-wide studies have made it possible to efficient- ly identify complex disease-associated genes. Using both the candidate gene approach and whole-genome linkage studies, 6 AITD susceptibility genes have been identified and confirmed; the first group includes the immunomodulatory gene products HLA-DR, CD40, cytotoxic T lymphocyte-associated factor (CTLA-4), and protein tyrosine phosphatase 22 (PTPN22), and the second group includes the thyroid-specific gene products thyroglobulin (Tg) and thyroid-stimulating hormone receptor (TSHR). Genetic factors predominate, accounting for approximately 80% of the likelihood of developing AITDs, whereas at least 20% is due to environmental factors (fig. 1). In recent years, a number of excellent reviews have been published on the genetic background of AITDs [13, 14].
An increased frequency of AITDs is reported in Turner syndrome (TS) and in other nondisjunctional chromosomal disorders such as Down and Klinefelter syndromes. The theory that maternal autoimmunity may lead to the preferential survival of a fetus with chromosomal aneuploidy is attractive but remains unproven [15]. The most prevalent autoimmune disorder in TS appears to be CAT, with a reported thyroid autoantibody incidence of 30– 50%. Hypothyroidism of autoimmune origin is so common in TS that almost every other TS woman will prob- ably develop hypothyroidism, and it increases with age [16, 17].
We know more about the minor details of AITDs, but the main question remains unanswered: why is the thyroid so prone to autoimmune disease? This review seeks to emphasize the role of the thyroid cell per se in AITDs and to focus attention on preventable exogenous factors.
Thyroid Cell Specificity
The thyroid cell produces a variety of immunologically active factors (table 1) and has complex nutrient requirements for hormonal synthesis and function (table 2), both of which influence susceptibility to AITDs. Thus, the thyroid cell is not just the innocent victim of an unchecked and disordered immune system. It is increasingly obvious that the target cells interact with the immune system, often in ways that seem defensive and protective, yet they can go awry and exacerbate autoimmunity under particular circumstances [11].

 In most human autoimmune diseases, the events that trigger autoimmunity remain elusive. Most importantly, it is unclear whether autoimmunity results primarily from an immune defect, is secondary to target organ alterations, or both. The thyroid shows increased iodine uptake and oxidation prior to lymphocytic infiltration concomitant with decreased thyroid epithelial cell proliferation in vitro. Modifying thyroid function influences the development of thyroid autoimmunity [18]. The thyroid cell, unlike other epithelial cells in the endocrine system, is unique because it releases hormonal products on its basal surface instead of its apical surface, thus allowing for the trafficking of precious iodine twice across the cell.
In most human autoimmune diseases, the events that trigger autoimmunity remain elusive. Most importantly, it is unclear whether autoimmunity results primarily from an immune defect, is secondary to target organ alterations, or both. The thyroid shows increased iodine uptake and oxidation prior to lymphocytic infiltration concomitant with decreased thyroid epithelial cell proliferation in vitro. Modifying thyroid function influences the development of thyroid autoimmunity [18]. The thyroid cell, unlike other epithelial cells in the endocrine system, is unique because it releases hormonal products on its basal surface instead of its apical surface, thus allowing for the trafficking of precious iodine twice across the cell.
Thyroid cells are capable of producing different factors (table 1), including IGF I, IGF II, and EGF, that can stimulate angiogenesis. The half-life of these molecules is short and they induce only local (non-systemic) effects. Stimulated thyroid follicular cells secrete several growth factors [19]. The expression of intercellular adhesion molecule-1 (ICAM-1) and lymphocyte function-associated antigen-3 (LFA-3) by thyroid cells is enhanced by IFN- , tumor necrosis factor (TNF), and interleukin (IL)-1. Thyroid cells express CD44, which acts as a homing receptor for hyaluronan, mediates leukocyte rolling (the first step in tissue homing), and may (like ICAM-1) induce lymphocyte activation under certain circumstances. Thyroid cells are now known to produce many cytokines (especially after stimulation with IL-1), including IL-1, IL-6, IL-8, IL-12, IL-13, and IL-15 [11]. Activated lymphocytes can produce TSH, which could have a variety of implications [20].
Low dose tolerance can easily be broken, and the thyroid is not well tolerated by the immune system. Auto- antigens in AITDs, as in other autoimmune endocrine diseases, include tissue-specific membrane receptors, enzymes, and secreted hormones. Mixed cellular and anti- body autoimmune responses are likely pathogenic to some degree. Circulating anti-Tg autoantibodies are also found in GD and CAT, as are autoantibodies to triiodothyronine (T3) and thyroxine (T4). The human (h) TSHR is the primary antigenic target in autoimmune hyperthyroidism [21]. The TPO autoantibody seems unlikely to have much pathogenic importance as it has limited access to TPO in vivo due to its location inside the cell. Further- more, anti-TPO autoantibodies do not inhibit the activity of the enzyme. Thus, their clinical value is principally to document thyroid gland autoimmunity. However, TPO may act as a hidden antigen because it is not adjacent to the vasculature.
In humans, excess thyroid hormone results in the attenuation of natural killer (NK) cell activity, which in theory could lead to the continuation of an autoimmune disorder. Upon return to a euthyroid status and the resulting normalization of NK activity, a reversion to control of the abnormal immune reaction would occur with perpetuation of GD. Additionally, an anti-idiotype might function as an agonist for the original antigen. Thus, an antibody to an antibody (anti-idiotype) to TSH might bind to the TSHR and stimulate the thyroid [22]. A more likely hypothesis is that anti-idiotypic antibodies are rarely produced at a detectable level. Hodkinson et al. [23] recently found a positive association between thyroid hormone concentration and NK-like T cells in the elderly. This relationship has not been investigated in young patients.
Antigen Presentation By The Thyroid Cell
Bottazzo et al. [24] first suggested that antigen presentation by HLA-DR-expressing thyroid cells may be a critical aspect of thyroid autoimmune disease. It quickly became apparent that the only stimulus able to induce MHC class II expression on thyroid cells was the T cell cytokine IFN- . Normal cells respond exactly the same as AITD thyroid cells to IFN- , and in animal models of AITDs class II expression on thyroid cells is always followed by the appearance of lymphocytes in the gland. In addition to inducing MHC class II expression, IFN- increases MHC class I expression on thyroid cells, thus allowing potential for the recognition of thyroid cells by cytotoxic CD8+ T cells [11].
It is possible that direct antigen presentation by the thyroid cell itself may occur in individuals who inherit thyroid-reactive T cells; such a circumstance would effectively bypass the classical macrophage-processing mechanism. The HLA-DR antigen-expressing thyroid cell may be as effective as the macrophage at presenting thyroid- specific antigens to the immune system [25], but the thyroid cell is incapable of supplying the costimulatory signals that professional antigen-presenting cells (APCs) do [11]. Any stimulus that causes increased DR expression on thyrocytes, such as IFN- produced by T cells in response to infection, combined with increased TSH stimulation may allow thyrocytes to function as APCs. Although thyroid cells may perform this function poorly, they are numerous and localized in one area, therefore allowing for increased production of the already established normally occurring low levels of antibodies [12].
Environmental Factors
A number of environmental factors have been implicated in the development of AITD in genetically predisposed individuals, including high iodine intake, Se deficiency, pollutants such as tobacco smoke, infectious dis- eases, certain drugs, and physical and emotional stress [26–30]. Herein, we focus on these preventable triggers. Individual susceptibility suggests that, in addition to genetics, some endogenous factors are also important to the development of AITDs, such as growth spurts in childhood, puberty, pregnancy, menopause, aging, and gender (fig. 1, 2).
 Iodine
Iodine
 Iodine
IodineDietary iodine plays an important role in the expression of AITDs. Epidemiological studies have suggested that AITDs are more common in areas of iodine sufficiency than in areas of iodine deficiency and that general increases in AITDs occur in parallel with increases in dietary iodine. CAT is less common in countries with a low iodine intake [27].
The thyroid requires the right amount of iodine. Either too much or too little causes problems. Too little io- dine brings all of the adaptive immune mechanisms of the thyroid into play, but despite these responses iodine deficiency disorders may still result. Too much iodine also affects the thyroid. Protective mechanisms include diminished trapping of iodide by the thyroid and de- creased iodide organification. In experimental thyroiditis several types of Tg epitopes have been found, including some containing iodine and/or hormones as well as some conformational epitopes. Experimentally increasing the iodination of Tg makes the protein more antigen- ic [28, 31]. Optimally, the iodine intake of a population should be kept within a relatively narrow interval that prevents iodine disorders, but not higher [29].
The mechanism of action of iodine in contributing to thyroid autoimmunity is not clear. Iodine may stimulate B lymphocytes to increase the production of immunoglobulin and thus induce AITDs by enhancing the activity of lymphocytes that have been primed by thyroid- specific antigens [30]. Iodine may enhance the antigen- presenting capabilities of macrophages, resulting in increased macrophage activity and enhanced lymphocyte stimulation. In addition, a high iodine intake in- creases the iodine content of the Tg molecule, which may increase its immunogenicity [31]. Lastly, iodine may provoke thyroid follicular cells to become APCs and thus potentiate AITDs by turning genetically predisposed normal thyrocytes into antigen-presenting thyrocytes.
Table 2 shows several minerals and trace elements that are essential for normal thyroid hormone metabolism. The role of these elements in childhood AITDs has not been well investigated.
Selenium
The second factor that has been strongly implicated in the development of autoimmune thyroiditis is the trace element Se. Se is a constituent of selenoproteins (SePs), in which it is incorporated as selenocysteine. Relevant actions of Se and SePs include antioxidant effects, appropriate functioning of the immune system, antiviral effects, influence on fertility, and a beneficial effect on mood [32]. Se deficiency is thought to be involved in the pathogenesis of autoimmune thyroiditis by lengthening the duration and exacerbating the severity of the disease; these effects may occur via reduced activity of the SeP glutathione peroxidase, which leads to an increased production of hydrogen peroxide. Another important class of SePs are the iodothyronine selenodeiodonases D1 and D2, which are responsible for producing biologically active T3 via 5 -deiodination in extrathyroidal tissues [33, 34].
Combined Se and iodine deficiencies lead to myxedematous cretinism. Adequate Se nutrition supports efficient thyroid hormone synthesis and metabolism and protects the thyroid gland from damage from excessive iodine exposure. In regions having severe combined deficiencies of iodine and Se, it is mandatory to normalize the Se supply before the initiation of iodine supplementation to prevent hypothyroidism [35].
In celiac disease, the inability to absorb Se may modulate SeP gene expression and promote intestinal mucosal damage, and this deficiency could additionally predispose to complications such as AITDs [34, 36].
Derumeaux et al. [37] discovered an inverse association between Se status and thyroid volume and echo- structure in French adults and concluded that Se may protect against AITDs. Duntas et al. [38] found beneficial effects when treating patients with autoimmune thyroiditis with selenomethionine for 6 months due to its ability to reduce anti-TPO antibodies. In the group treat- ed with LT4 combined with Se, these effects were very prominent in the first 3 months and were further sustained after 6 months of treatment. A striking majority of the patients reported an improvement in mood and well-being.
Environmental Pollutants
Various environmental toxins and pollutants have been implicated in the induction of AITDs.
Polyhalogenated biphenyls are commonly used com- pounds with a wide variety of industrial applications. Polybrominated biphenyls are flame retardant, and polychlorinated biphenyls (PCBs) are used as lubricants, adhesives, inks, and plasticizers. PCBs are known to accumulate in lakes and rivers and subsequently in the adipose tissue of fish and humans [27]. These compounds might trigger AITDs by interfering with iodide transport and inducing oxidative stress. There is evidence that peri- natal PCB exposure decreases thyroid hormone levels in rat pups. In adults, adolescents, and children from highly PCB-exposed areas, the concentration of PCBs in blood samples negatively correlated with levels of circulating thyroid hormones [39, 40]. Populations with long-term exposure to PCBs have increased prevalences of anti-TPO antibodies, which is probably related to the immunomodulatory effects of PCBs. Pollutants from car emissions and heavy industry as well as coal pollution and agricultural fungicides are also implicated in AITD development [26, 27].
Smoking is associated with an increased risk of developing GD and with a reduced remission rate after thionamide treatment. Even more striking is the effect of smoking on Graves’ orbitopathy, which tends to be more severe in smokers [32, 41]. Smoking might contribute to the pathogenesis of GD by altering the structure of the thyrotropin receptor, making it more immunogenic and leading to the production of thyrotropin receptor-stimulating antibodies that react strongly with retroorbital tissue [41]. Smoking induces the polyclonal activation of B and T cells and increases presentation of antigens by damaged cells. Hypoxia may play a role in Graves’ orbitopathy because retrobulbar fibroblasts show a significant increase in proliferation and glycosaminoglycan production when cultured under hypoxic conditions [42, 43]. The effects of parental smoking on thyroid function in fetuses or 1-year-old infants [44] provide additional insight into the interrelationship between smoking and thyroid dysfunction. The latter study found that infants whose mothers and fathers smoked had higher cord serum concentrations of Tg and thiocyanate than did infants whose parents did not smoke. The clinical picture observed in adolescents exposed to passive smoking could be due to direct stimulation of sympathetic nervous activity by nicotine in addition to the smoking-induced increase in thyroid hormone secretion [45].
The association of smoking with CAT is less well defined. Although a relationship with autoimmune hypothyroidism or postpartum thyroiditis has been reported, this finding was not supported by meta-analysis of the published papers [32, 45].
Infections
In some individuals, autoimmunity is the price that must be paid for the eradication of an infectious agent. Infections have been implicated in the pathogenesis of several autoimmune, endocrine, and non-endocrine diseases. Either viral or bacterial infections might represent a risk factor for the development of AITDs. Viruses have long been suspected as etiological agents in many auto- immune diseases, including AITDs; moreover, a viral cause of AITDs, infecting either the thyroid or immune cells, has been demonstrated in an avian model. Although viruses may be likely etiological agents in human AITDs, this possibility remains unproven [25, 27, 30].
An increased frequency of antibodies to the influenza B virus has been observed in a group of patients with thyrotoxicosis. In addition, virus-like particles have been found in the thyroids of chickens with autoimmune thyroiditis, with similar particles detected in the thyroids of humans. Serological evidence of prior staphylococcal and streptococcal illnesses has been described in a few patients with AITDs [27].
Some of the strongest evidence linking infectious agents to the induction of AITDs has been the association of Yersinia enterocolitica infection with thyroid disease. This Gram-negative coccobacillus commonly causes diarrhea along with a variety of abnormalities that suggest autoimmune disease, including arthralgias, arthritis, erythema nodosum, carditis, glomerulonephritis, and iritis. Weiss et al. [46] demonstrated that Y. enterocolitica had a saturable, hormone-specific binding site for the mammalian TSH that resembled the receptor for TSH in the human thyroid gland.
An immune response against a viral antigen that shares homology with the TSHR may be the inductive event that ultimately leads to TSHR autoimmunity [21]. A significant association between hepatitis C and AITDs has been found. Anti-TPO antibody titers have been shown to increase at the end of treatment with IFN- in patients with the hepatitis C virus, and these patients were more susceptible to AITDs than were hepatitis B patients. These patients should be screened for autoimmune thyroiditis before and after IFN treatment [47, 48].
Infection might induce an autoimmune response by various mechanisms, such as molecular mimicry, polyclonal T cell activation by microbial superantigens, and increased thyroid expression of human leukocyte anti- gens [49]. Inflammation induced by viral infections or by pollutants can modify cell signaling pathways and influence T cell activity and cytokine secretion profiles [26].
Drugs
Several drugs have been implicated in the pathogenesis of AITDs. Amiodarone is an iodine-containing drug with diverse effects on thyroid function. Serum titers of TPO antibodies are elevated in approximately half of the patients who develop amiodarone-induced hypothyroid- ism. Amiodarone has also been shown to affect T cell function [27]. Thyroid antibodies disappeared from the circulation 6 months after amiodarone discontinuation [32].
Lithium, a psychopharmaceutical and well-known goitrogen, has been shown to inhibit thyroid hormone release. Antithyroid antibodies are found more frequently in psychiatric patients on lithium therapy than in similar psychiatric patients treated with other drugs. Lithium-induced increases in serum TSH concentrations might enhance autoantigen expression on the surface of thyrocytes, thereby exacerbating autoimmune responses [32, 50].
Other agents involved in thyroid autoimmunity are IL-2 (thyroid autoimmune phenomena with or without hypothyroidism), IFN- (thyroid dysfunction, hypothyroidism, and occurrence of thyroid autoantibodies), highly active antiretroviral therapy (HAART; possible occurrence of thyroid autoimmune phenomena and dysfunction), and Campath-1H, a humanized monoclonal antibody targeting the CD52 antigen on lymphocytes and monocytes that is used after transplantation (occurrence of GD) [32].
Stress
Although numerous anecdotal reports have associated the onset of AITDs, and particularly GD, with stressful events, objective evidence has been difficult to obtain. Both psychological stress, such as bereavement, and physical stress, such as trauma or major illness, have been implicated [27].
Neuroendocrine immune mechanisms responsible for the putative effects of stress on the onset and course of GD are poorly defined, but they might include activation of the HPA axis (although this should cause immunosuppression) and a shift from a Th1 (cell-mediated) immune response to a Th2 (humoral) immune response [32, 51].
Additionally, heat shock proteins (HSPs), which are well-known stress proteins, could share epitopes with the TSHR. Heufelder et al. [52] found that high levels of HSP- 72 expression in AITDs may reflect a state of chronic cellular stress, but this finding could also indicate an immunomodulatory function of HSP-72 in AITDs. HSPs are ubiquitous, highly conserved proteins that are expressed in response to a wide variety of physiological and environmental insults. They allow cells to survive otherwise lethal conditions. HSPs have been postulated to be critical antigens in both autoimmune diseases and experimental models of autoimmunity [53, 54].
Improving stress by the prolonged use of bromazepam has been shown to increase the remission rate of hyper- thyroidism after a thionamide course [55]. The relation- ship between stress and CAT is less evident. Graves’ patients might be stressed because of hyperthyroidism and not hyperthyroid because of stress, whereas CAT patients are not stressed because they are euthyroid or hypothyroid [32]. Whatever the mechanism of action, stress may cause decompensation in a genetically susceptible individual and lead to the induction or exacerbation of an AITD.
Pregnancy And Postpartum
AITDs tend to be more frequent in women. The reason for this gender-related difference is not clear and is not explained by the additional X chromosome in females [42]. The possibility that genes responsible for immune responses are located on the X chromosome has been considered but not confirmed. Sex steroids could modify immune responses by acting directly on immune cells. Estrogens are well-known stimulators of TSH secretion, which could enhance HLA-DR expression. Parity per se does not seem to play a significant role [32, 56].
The accumulation of fetal cells in the maternal thyroid gland during pregnancy (painless postpartum thyroiditis) may induce autoimmune thyroiditis [57]. Pregnancy is accompanied by a suppression of the immune system with a shift in the Th1/Th2 balance towards Th2 immunity, a process that is aimed at protecting the fetus. A possible link between pregnancy and the postpartum occurrence of AITDs might be represented by fetal microchimerism. Fetal cells pass into the maternal circulation and may persist in the maternal blood. Microchimerism of presumed fetal origin has been shown in thyroid tissue specimens of women with previous pregnancies, particularly in those with AITDs. The persistence of activated intrathyroidal fetal cells might influence thyroid autoimmunity in genetically susceptible women by modulating or even initiating maternal immune responses in a graft- versus-host reaction upon termination of pregnancy-re- lated immune suppression. It cannot presently be ruled out, however, that intrathyroidal fetal cells are only innocent bystanders and do not participate in triggering or exacerbating thyroid autoimmune responses [32, 54, 58]. Mothers who have given birth to sons have thyroidal Y chromosome-positive cells more frequently if they are affected by either CAT or GD than if they have thyroid adenomas [59].
The presence of elevated TPO antibodies in about 10% of pregnant women is associated with an increased risk of miscarriage, gestational thyroid dysfunction, and postpartum thyroiditis [48]. Maternal-to-fetal transfer of TSHR antibodies with polyclonal activity and a different half-life can lead to a transient perinatal thyroid dysfunction, opposite to a maternal one [60].
Conclusion
A rapidly growing body of evidence on the interplay between genetic, environmental, and endogenous factors has expanded our knowledge of the complex etiopathogenesis of AITDs. Autoimmune thyroid disorders are examples of common diseases in which immunogenetic factors play an important role.
The thyroid cell itself appears to play a major role in disease progression by interacting with the immune system. The complexity of hormonal synthesis, unique oligoelement requirements, and the specific capabilities of the thyroid cell defense system probably make the thyroid prone to AITDs. The initial insult to the human thyroid gland that activates the onset of AITDs remains un- known and seems to be strongly individual. Understand- ing more about the interaction between genes and the environment could yield entirely novel pathways, some of which might be as simple as identifying the need to avoid smoking or to control the intake of particular nutrients. Evidence for many causal agents is, however, scarce, and more data are certainly required. We believe that it is particularly important to draw attention to this problem in pediatric patients. Lessons learned from the enigmatic questions raised in AITD studies could clarify the pathogenesis of other organ-specific autoimmune disorders.
L. Saranac S. Zivanovic B. Bjelakovic H. Stamenkovic M. Novak B. Kamenov Pediatric Clinic, University Clinical Center, Nis, Serbia
1 Phillips DI, Osmond C, Baird J, Huckle A,
Rees-Smith B: Is birthweight associated with
thyroid autoimmunity? A study in twins.
Thyroid 2002;12:377–380.
2 Brix TH, Hansen PS, Rudbeck AB, Hansen
JB, Skythe A, Kyvik KO, Hegedus L: Low
birth weight is not associated with thyroid
autoimmunity: a population-based twin
study. J Clin Endocrinol Metab 2006;91:
3499–3502.
3 Wilkin TJ: The great weight gain experiment,
accelerators and their implications for
autoantibodies in diabetes. Arch Dis Child
2006;91:456–458.
4 Matarese G, La Cava A, Sanna V, Lord MG,
Lechler RI, Fontana S, Zappacosta S: Balancing
susceptibility to infection and autoimmunity:
a role of leptin? Trends Immunol
2002;23:182–187.
5 Radetti G, Kleon W, Buzi F, Crivellero C,
Pappalardo L, Di Lorgi N, Maghnie M: Thyroid
structure and function are affected in
childhood obesity. J Clin Endocrinol Metab
2008;93:4749–4754.
6 Pacifico L, Di Renzo L, Anania C, Osborn JF,
Ippoliti F, Schiavo E, Chiesa C: Increased Thelper
interferon-gamma-secreting cells in
obese children. Eur J Endocrinol 2006;154:
691–697.
7 Marras V, Casini MR, Pilia S, Carta D, Civolani
P, Porcu M, Uccheddu AP, Loche S: Thyroid
function in obese children and adolescents.
Horm Res Paediatr 2010;73:193–197.
8 Saranac L, Zivanovic S, Novak M: High fT3
(free triiodothyronine), new syndrome or innocent
bystander. Endocr Abstracts Eur
Congr Endocrinol, Prague, 2010, p 771.
9 Schwartz M, Cohen IR: Autoimmunity can
benefit self-maintenance. Immunol Today
2000;21:265–268.
10 Cohen IR, Schwartz M: Autoimmune maintenance
and neuroprotection of the central
nervous system. J Neuroimmunol 1999;100:
111–114.
11 Weetman AP: Autoimmune thyroid disease:
propagation and progression. Eur J Endocrinol
2003;148:1–9.
12 Weetman AP: New aspects of thyroid autoimmunity.
Horm Res 1997;48(suppl 4):51–
54.
13 Jacobson EM, Tomer Y: The CD40, CTLA-4,
thyroglobulin, TSH receptor, and PTPN22
gene quintet and its contribution to thyroid
autoimmunity: back to the future. J Autoimmun
2007;28:85–98.
14 Tomer Y, Huber A: The etiology of autoimmune
thyroid disease: a story of genes and
environment. J Autoimmun 2009;32:231–
239.
15 Saenger P: Turner syndrome; in Sperling MA
(ed): Pediatric Endocrinology, ed 3. Philadelphia,
Saunders Elsevier, 2008, pp 610–661.
16 El-Mansoury M, Bryman I, Berntorp K,
Hanson C, Wilhelmsen L, Landin-Wilhelmsen
K: Hypothyroidism is common in Turner
syndrome: results of a five-year follow up.
J Clin Endocrinol Metab 2005;90:2131–2135.
17 Mortensen KH, Cleemann L, Hjerrild BE,
Nexo E, Locht H, Jeppesen EM, Gravholt
CH: Increased prevalence of autoimmunity
in Turner syndrome – influence of age. Clin
Experim Immunol 2009;156:205–210.
18 Homo-Delarche F, Boitard C: Autoimmune
diabetes: the role of the islets of Langerhans.
Immunol Today 1996;17:456–460.
19 Denef JF, Ovaert C, Many MC: Experimental
goitrogenesis (in French). Ann Endocrinol
(Paris) 1989;50:1–15.
20 Fabry Z, Raine CS, Hart MN: Nervous tissue
as an immune compartment: the dialect of
the immune response in the CNS. Immunol
Today 1994;15:218–224.
21 Song YH, Li Y, Maclaren NK: The nature of
autoantigens targeted in autoimmune endocrine
diseases. Immunol Today 1996;17:232–
238.
22 Zakarija M, McKenzie JM: The spectrum
and significance of autoantibodies reacting
with the thyrotropin receptor. Endocrinol
Metab Clin North Am 1987;16:343–364.
23 Hodkinson CF, Simpson EEA, Beattie JH,
O’Conor JM, Campbell DJ, Strain JJ, Wallace
JM: Preliminary evidence of immune function
modulation by thyroid hormones in
healthy men and women aged 55–70 years. J
Endocrinol 2009;202:55–63.
24 Botazzo GF, Pujol-Borrell R, Hanafusa T,
Feldmann M: Role of aberrant HLA-DR expression
and antigen presentation in induction
of endocrine autoimmunity. Lancet
1983;2:1115–1119.
25 Davies TF, Piccini LA: Intrathyroidal MHC
class II antigen expression and thyroid autoimmunity.
Endocrinol Metab Clin North
Am 1987;16:247–268.
26 Duntas LH: Environmental factors and autoimmune
thyroiditis. Nat Clin Pract Endocrinol
Metab 2008;4:454–460.
27 Safran M, Paul TL, Roti E, Braverman LE:
Environmental factors affecting autoimmune
thyroid disease. Endocrinol Metab
Clin North Am 1987;6:327–342.
28 Dunn JT: What is happening with our iodine?
J Clin Endocrinol Metab 1998;3398–
3400.
29 Laurberg P, Cerqueira C, Ovesen L, Rasmusen
LB, Perrild H, Andersen S, Pedersen IB,
Carle A: Iodine intake as a determinant of
thyroid disorders in population. Best Pract
Res Clin Endocrinol Metab 2010;24:13–27.
30 Weetman AP, McGregor AM: Autoimmune
thyroid disease: developments in our understanding.
Endocr Rev 1984;5:309–355.
31 Carayanniotis G, Rao VP: Searching for
pathogenic epitopes in thyroglobulin: parameters
and caveats. Immunol Today 1997;
18:83–88.
32 Bartalena L, Tanda ML, Piantanida E, Lai A,
Compri E, Lombardi V: Environment and
thyroid autoimmunity; in Wiersinga WM,
Drexhage HA, Weetman AP, et al (eds): The
Thyroid and Autoimmunity: Merck European
Thyroid Symposium Noordwijk 2006,
June 15–18. Stuttgart, Thieme, 2007 pp 60–
73.
33 Berry MJ, Bany L, Larsen PR: Type I iodothyronine
deiodinase is a selenocysteine-containing
enzyme. Nature 1991;349:438–440.
34 Duntas LH: Selenium and inflammation:
underlying anti-inflammatory mechanisms.
Horm Metab Res 2009;41:443–447.
35 Zimmerman MB, Kohrle J: The impact of
iron and selenium deficiencies on iodine and
thyroid metabolism: biochemistry and relevance
to public health. Thyroid 2002;12:
867–878.
36 Duntas LH: Does celiac disease trigger autoimmune
thyroiditis. Nat Rev Endocrinol
2009;5:190–191.
37 Derumeaux E, Valeix P, Castetbon K, Bensimon
M, Boutron-Ruault MC, Arnaud JH,
Hercberg S: Association of selenium with
thyroid volume and echostructure in 35- to
60-year-old French adults. Eur J Endocrinol
2003;148:309–315.
38 Duntas LH, Mantzou E, Koutras DA: Effects
of a six month treatment with selenomethionine
in patients with autoimmune thyroiditis.
Eur J Endocrinol 2003;148:389–393.
39 Meerts IA, Assink Y, Cenijn PH, Van Den
Berg JH, Weijers BM, Bergman A, Koeman
JH, Brouwer A: Placental transfer of a hydroxylated
polychlorinated biphenyl and effects
on fetal and maternal thyroid hormone
homeostasis in the rat. Toxicol Sci 2002;68:
361–372.
40 Boas M, Feldt-Rasmussen U, Skakkebaek
NE, Main KM: Environmental chemicals
and thyroid function. Eur J Endocrinol 2006;
154:599–611.
41 Utiger RD: Effects of smoking on thyroid
function. Eur J Endocrinol 1998;138:368–
369.
42 Prummel MF, Strieder T, Wiersinga WM:
The environment and autoimmune diseases.
Eur J Endocrinol 2004;150:605–618.
43 Pontikides N, Krassas GE: Influence of cigarette
smoking on thyroid function, goiter
formation and autoimmune thyroid disorders.
Hormones (Athens) 2002;1:91–98.
44 Gasparoni A, Autelli M, Ravagni-Probizer
MF, Bartoli A, Regazzi-Bonora M, Chirico
G, Rondini G: Effect of passive smoking on
thyroid function in infants. Eur J Endocrinol
1998;138:379–382.
45 Vestergaard P: Smoking and thyroid disorders
– a meta-analysis. Eur J Endocrinol
2002;146:153–161.
46 Weiss M, Ingbar SH, Winblad S, Kasper DL:
Demonstration of a saturable binding site for
thyrotropin in Yersinia enterocolitica . Science
1983;219:1331–1333.
47 Fernandez-Soto L, Gonzales A, Escobar-Jimenez
F, Vazquez R, Ocete E, Olea N, Salmeron
J: Increased risk of autoimmune thyroid
disease in hepatitis C vs B before, during and
after discontinuing interferon therapy. Arch
Intern Med 1998;158:1445–1448.
48 Testa A, Castaldi P, Fanti V, Fiore GF, Grieco
V, De Rosa A, Pazardjklian MG, De Rosa G:
Prevalence of HCV antibodies in autoimmune
thyroid disease. Eur Rev Med Pharmacol
Sci 2006;10:183–186.
49 Davies TF: Infection and autoimmune thyroid
disease. J Clin Enocrinol Metab 2008;93:
674–676.
50 Lazarus JH, John R, Bennie EH, Chalmers
RJ, Crockett G: Lithium therapy and thyroid
function: a long-term study. Psychol Med
1981;11:85–92.
51 Dayan CM: Stressful life events and Graves’
disease revisited. Clin Endocrinol (Oxf)
2001;55:13–14.
52 Heufelder AE, Goellner JR, Wenzel BE,
Bahn RS: Immunohistochemical detection
and localization of a 72-kilodalton heat
shock protein in autoimmune thyroid disease.
J Clin Endocrinol Metab 1992;74:724–
731.
53 Parcellier A, Gurbuxani S, Schmitt E, Solary
E, Garrido C: Heat shock proteins, cellular
chaperones that modulates mitochondrial
cell death pathways. Biochem Biophys Res
Commun 2003;304:505–512.
54 Gaston JS: Are heat shock proteins involved
in autoimmunity? Int Clin Lab Res 1992;22:
90–94.
55 Benvenga S: Benzodiazepine and remission
of Graves’ disease. Thyroid 1996;6:659–660.
56 Adams D: How the immune system works
and why it causes autoimmune diseases. Immunol
Today 1998;17:300–303.
57 Pierce EN, Farwel AP, Braverman LE: Thyroiditis.
N Engl J Med 2003;348:2646–2655.
58 Badenhoop K: Microchimerism and the
model of postpartum thyroiditis; in Wiersinga
WM, Drexhage HA, Weetman AP, et
al (eds): The Thyroid and Autoimmunity:
Merck European Thyroid Symposium
Noordwijk 2006, June 15–18. Stuttgart,
Thieme, 2007, pp 99–103.
59 Szabolcs I: Clinical relevance of thyroid peroxidase
autoantibodies in euthyroid individuals;
in Wiersinga WM, Drexhage HA,
Weetman AP, et al (eds): The Thyroid and
Autoimmunity: Merck European Thyroid
Symposium Noordwijk 2006, June 15–18.
Stuttgart, Thieme, 2007, pp 133–142.
60 Saranac L, Miljkovic M, Stamenkovic H, Mileusnic-Milenovic
R, Petrovic G, Kamenov
B: Late onset transient thyroid dysfunction
in children born to mothers with autoimmune
thyroid disease. Facta Univ Ser Med
Biol 2003;10:52–56.
Post Disclaimers
General Disclaimer, Licenses and Board Certifications *
Professional Scope of Practice *
The information herein on "Why Is The Thyroid So Prone To Autoimmune Disease?" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and on our family practice-based chiromed.com site, focusing on naturally restoring health for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine; wellness; contributing etiological viscerosomatic disturbances within clinical presentations; associated somato-visceral reflex clinical dynamics; subluxation complexes; sensitive health issues; and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and licensure jurisdiction. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that directly or indirectly relate to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933





Comments are closed.